The DNA Network |
| Self Assembly: Wikification [business|bytes|genes|molecules] Posted: 07 Sep 2008 07:00 PM CDT
One of my goals for 2008 had been to move away from my current shared hosting to more virtualized hosting and development environments, which is why I got rid of my desktops, and am now using my macbook pro, a slice on Slicehost and Amazon Web Services (quite literally eating my own dogfood) as my computing/hosting environment. About half way there. In the past I had written about adopting Wikispaces as a wiki platform. I still love the platform, but things I wanted to do required hosting the wiki myself, and that led me to MindTouch Deki. It’s not quite your traditional wiki, but fits very well into what I want to use it for. That move coincided with the move to Slicehost, where the the bbgm wiki was the first thing that was set up. It’s taken a year to really get things to a point where the original idea makes sense, but slow and (not so) steady wins the race. The original germ came from Tantek Çelik’s Wiki. Over time I hope the bbgm wiki becomes its own resource, a place that contains some of the research and ideas behind the blog as well as various things I learn from attempts at doing some science and bad programming. The Deki platform has a RESTful API, so it can be used as more than just a content repository. Also the commenting functionality makes it possible to get feedback. If anyone is interested in contributing/editing/modifying, please let me know and I will add you as authors. I’ve also started funneling a feed of recent changes into Friendfeed. Next step - move bbgm to Slicehost and do more with deepaksingh.net>/a> Related articles by Zemanta
![Reblog this post [with Zemanta]](http://img.zemanta.com/reblog_b.png?x-id=751ee0a8-4f21-4055-96eb-fcb278c22531) |
| The Davis Food Coop Reviews Tomorrow's Table [Tomorrow's Table] Posted: 07 Sep 2008 06:46 PM CDT The Davis Food Coop, our local grocery store, has come out with a review of Tomorrow's Table. Here is our response to the review: A local, fresh perspective on genetic engineering and organic farming Our existing agricultural system, while productive, has serious problems that negatively effect the environment and it's inhabitants. These problems are caused by the overuse of pesticides, synthetic fertilizers, and farming practices that lead to soil erosion. A major goal of sustainable agriculture is to greatly reduce or eliminate these problems while maintaining yields and farm incomes. In our book, Tomorrow's Table: Organic Farming, Genetics, and the Future of Food, we suggest a few essential ideas to help forge a more sustainable agriculture. We advocate adopting technologies or farming practices that: Produce abundant, safe and nutritious food Reduce harmful environmental inputs Provide healthful conditions for farm workers Protect the genetic make-up of native species Enhance crop genetic diversity Foster soil fertility Improve the lives of the poor and malnourished Maintain the economic viability of farmers and rural communities Not surprisingly, given our expertise, we believe that organic farming and genetic engineering each have something to contribute to a sustainable agriculture. Rather than embracing "GE crops as the unqualified answer" as Miller states in her review of our book, we advocate that each new approach be evaluated on a case-by-case basis in light of these criteria. An appropriate technology for food and farming, as asserted by the economist Schumacher in his book Small is Beautiful, should promote health, beauty, and permanence. It should be low cost and low maintenance. Considering Schumacher's ideas and our goals for ecological farming, it is apparent that GE will sometimes be appropriate for crop production and sometimes not. This is because GE is simply a tool that can be applied to a multitude of uses, depending on the needs of farmers, and consumers. Still, as we attempt to show in our book, GE comprises many of the properties advocated by Schumacher. It is a relatively simple technology that scientists in most countries, including many developing countries, have perfected. The product of GE technology, a seed, requires no extra maintenance or additional farming skills. GE seeds can be saved and then passed down from generation to generation and improved along the way. It is therefore clear that humans will likely reap many significant and life-saving benefits from GE. This is because even incremental increases in the nutritional content, disease resistance, yield, or stress tolerance of crops can go a long way to enhancing the health and well-being of farmers and their families. Applications of GE have already been used to reduce the adverse environmental effects of farming and enable farmers to produce and sell more food locally. For example, when small-scale papaya farmers in Hawaii were confronted with a devastating viral disease, GE papaya was the most appropriate approach (funded by non-profit sources and distributed free to growers) to restore the industry. There were no conventional or organic methods to control the disease then, nor are there now. GE crops in combination with organic techniques have already helped farmers in less developed countries. For example flooding is a major problem for millions of farmers that live on less than a dollar a day in Bangladesh, and India. Yet for over 50 years, breeders were unsuccessful in developing flood-resistant rice using conventional breeding. Today, using advanced genetic techniques, we (Pam and her colleagues) have been able to produce such a variety that has been embraced by growers because of its 2-5 fold higher yield in flood zones. Scientists predict that the lives of thousands of children dying from vitamin-A deficiency will be saved once GE rice fortified with precursors to vitamin A (so-called "Golden Rice") is released in 2011. The best way to determine if practices are effective is through scientific study and peer review. Trying to evaluate agricultural technology without peer-reviewed science is like trying to determine if there are weapons of mass destruction in Iraq without inspections. When scientific information is available, we should use it. For example we now know that the introduction of GE cotton has dramatically reduced the use of insecticides in the US and abroad. In fields where the GE cotton is not used, the scientific data on the effects of chemical insecticides on insect biodiversity are unequivocal; they devastate local populations. In regards to eating GE foods currently on the market, the overall issue is health. We would be quite concerned if genes in GE crops could harm people. But this is not the case. There is broad scientific consensus that the GE crops on the market are safe to eat. Over the last 15 years, 1 billion acres have been planted and not a single instance of harm to human health or the environment has been documented. In contrast, each year tens of thousands of people are poisoned by pesticides. Agricultural advances need to be shared globally. The oft-repeated idea that because we have an abundance of food to eat in the US (thanks to good soils and abundant water and advances made by geneticists, farmers and breeders), we don't need to continue to improve crops in other countries is short-sighted. It doesn't make sense for the US to grow food and ship it to Africa or S. Asia where people cannot afford to buy it. Plus it takes precious energy to move it. Farmers in less developed countries need their own local production, improved seed, farming practices and sound government policies. That way they can feed themselves, just as we do here. Pitting genetic engineering and organic farming against each other only prevents the transformative changes needed on our farms. Rather than opposing all applications of a particular technology, lets direct the technology to help forge a sustainable agriculture. In the words Rachel Carson, author of Silent Spring (1962): "A truly extraordinary variety of alternatives to the chemical control of insects is available. Some are already in use and have achieved brilliant success. Others are in the stage of laboratory testing. Still others are little more than ideas in the minds of imaginative scientists, waiting for the opportunity to put them to the test. All have this in common: they are biological solutions, based on understanding of the living organisms they seek to control, and of the whole fabric of life to which these organisms belong. Specialists representing various areas of the vast field of biology are contributing—entomologists, pathologists, geneticists, physiologists, biochemists, ecologists—all pouring their knowledge and their creative inspirations into the formation of a new science of biotic controls." Pam and Raoul, Davis Food Coop shareholders since 1980 "Tomorrow's Table" is now available in the coop. To view peer-reviewed citations, learn more about GE and organic farming, to see other reviews of the book, or to continue this dialog, please check out Pam's blog at http://pamelaronald.blogspot.com |
| 10 Things about GE crops to Scratch From Your Worry List [Tomorrow's Table] Posted: 07 Sep 2008 08:17 AM CDT As New York Times columnist John Tierney points out in a recent article, many consumers worry about things that are not actually a threat to human health or to the environment. This is especially true in the case of food, farming and genetic engineering. While there are plenty of issues to debate (how can we make the best seed available to farmers that need it; how can we conserve land, how can we have productive farms without harming the environment), there are some scientific facts that have not yet seeped into the public consciousness. With September upon us, perhaps you're in the mood for a break, so I've rounded up a list of 10 things not to worry about this fall. You can make your own nominations on this blog. 1. GE crops require more pesticides Two classes of pesticides are affected by GE crops: Herbicides that kill weeds and insecticides that kill insects. There is now clear and ample evidence that BT crops have reduced the use of insecticides- here and abroad. In China, cotton farmers were able to eliminate 150 million pounds of insecticide in a single year by using GE varieties. For comparison, in California, we spray 190 million pounds each year. Although BT-cotton in China has been dramatically effective in reducing pesticide use, after 7 years some farmers started spraying again to control secondary insect pests that are not controlled by Bt. This points to the need to integrate GE crops into organic farming systems that also use crop rotation and beneficial insects to control secondary pests. In India a huge proportion of the farming expenses are going to insecticides. Although GE seed are more expensive, the yields are 80% higher and the farmer saves on insecticide costs. In the US, pesticide use on corn, soybeans, and cotton declined by about 2.5 million pounds in the United States since the introduction of GE crops in 1996 (Fernandez-Cornejo and Caswell 2006). In the case of herbicides applications, usage per acre has declined since the advent of herbicide-resistant crops (Fernandez-Cornejo and Caswell 2006), and because glyphosate breaks down quickly in the environment, the overall net effect is a reduction in the toxicity of herbicides used. For example, conventional soybean growers used to apply the more toxic herbicide metolachlor to control weeds of soybeans despite the fact that metolachlor is a known groundwater contaminant and is included in a class of herbicides with suspected toxicological problems. Switching from metolachlor to glyphosate in soybean production has had huge environmental benefits not measured in pounds of active ingredient but in environmental impact (Fernandez-Cornejo and McBride 2002). Another agricultural benefit is that herbicide-resistant soybean has helped foster use of low-till and no-till agriculture, which leaves the fertile topsoil intact and protects it from being removed by wind or rain. Also, because tractor-tilling is minimized, less fuel is consumed and greenhouse gas emissions are reduced (Farrell et al. 2006). 2. Corporate control of GE seeds forces farmers to buy seed each year Most conventional and organic farmers in California buy their seed. They are not contractually obligated to buy seed from the same company each year (this is the same for GE and conventional seed). Farmers choose to buy hybrid seed and other improved seed because they are higher yielding and/or have qualities preferred by consumers. Although the US seed industry is dominated by large corporations, this was the case before GE came into play. In some less developed countries, such as Bangladesh, national breeding programs distribute seed (GE or conventional) freely to farmers who can then self their own seed. For example a flood-tolerant gene cloned in my lab and used to develop new varieties in collaboration with colleagues has been distributed to Bangladeshi farmers through national breeding programs. The farmers are now saving the seed to share with neighbors. In China and India, there are burgeoning seed industries and an increasing fraction of the agricultural seed is purchased, not provided free by the government. For example, over half of the rice planted now in China is hybrid, therefore purchased annually, most of it produced by private companies. (Thanks to Kent Bradford for providing this information). 3. If we could just distribute the food available, impoverished people would not need GE seed. Many rural poor in Africa and S. Asia cannot afford to buy food. They must grow their own. Rather than relying on shipments from abroad, they need help improving their own production practices. This includes access to improved seed, farming practices and sound government policies. Now that is something we do need to worry about. In China, there are proven health benefits to families because of the reduction in pesticide use after the introduction of GE crops. A recent analysis predicts that the lives of thousands of children will be saved once golden rice is launched in 2011. 4. We don't need GE to improve seed, we can use marker-assisted breeding. Marker assisted breeding is a hybrid of conventional breeding and GE that relies on modern genetic techniques to modify seed. Flood tolerant rice, developed through marker assisted breeding, has the potential assist 75 million farmers who live on less than a dollar a day in major flood zones in places like Myanmar, Bangladesh, and India. There are other agricultural problems, however, that cannot be addressed using marker-assisted breeding. For example, in the 1990s, papaya orchards on the island of Hawaii were threatened by papaya ring spot virus. Dennis Gonsalves, a former plant virologist at Cornell who is now with the U.S. Department of Agriculture, developed a genetically engineered papaya variety that was completely resistant to the virus. The GE papaya yielded 20 times more than the conventional variety was was distributed freely to growers. It's a great example of genetic engineering benefiting local farmers. There was no other tool available to combat papaya ringspot virus, nor is there now. 5. GE crops harm human health. There is virtually universal scientific consensus that GE crops currently on the market are safe to eat. After over 10 years of consumption there has not been a single validated report of negative health effects from any GE crop. In contrast every year there are thousands of reported pesticide poisonings (ca. 1200 each year in California alone). Every new GE crop must be evaluated on a case-by-case basis. Lets direct our efforts to generating new crop varieties that will benefit the maximum number of people. 6. GE crops harm the environment GE crops themselves have had no negative environmental effects after over 1 billion acres have been planted. The rising problem of herbicide resistant weeds and its consequences, for example, are a significant environmental (and practical) problem. But this is a problem anytime an herbicide is used extensively and is not an environmental consequence of the GE crop itself. The same result would occur if the crop is GE or non-GE (and many such herbicide tolerant non-GE crops have been generated). Like non-GE crops, GE crops produce pollen. Thus pollen flow from GE crops poses essentially the same risks as non-GE crops. There is no example of a domesticated crops escaping into the wild and wreaking environmental harm. This is because they are highly domesticated and need a farmers care to survive. As Freeman Dyson once said, "Have you seen any wild poodles lately?" A positive spin off of the whole GE debate is that now there is more attention being placed on the ecological effects of pollen flow from domesticated crops to wild populations. 7. GE crops reduce biological diversity Bt has had some real benefits in reducing chemical insecticide use and in enhancing insect biodiversity. The recent analysis by Cattaneo et al (2005) clearly shows that there is similar biodiversity (ants and beetles) in GE cotton fields vs non-GE cotton fields. In contrast, broad-spectrum insecticides (which are used on the vast majority of non-GE cotton fields throughout the world) significantly reduces ant and beetle species richness. If we hadn't genetically modified our crops by conventional methods over the last 50 years, we would be using twice as much of the Earth's surface to grow the same amount of food. In the future, if we don't increase yields, we'll need to use double the amount of land to produce the same amount of food. Sparing land from becoming farmland, is the greatest benefit to biodiversity. For this reason, some ecologists see the application of GE as a way to spare even more land from destruction by enhancing yields (Qaim and Zilberman 2003; Snow et al. 2005). 8. The regulation of GE crops is lax. GE crops are more stringently regulated than other crops. That is, other crops are not regulated at all. Hundreds of thousands of children are dying each year because of vitamin A deficiency, thousands more are poisoned with pesticides, more land is being put into production every year, which negatively affects global warming. In contrast there has not been a single case of harm from GE crops to human health or the environment, even after over 1 billion acres grown. Their is broad scientific consensus that the GE crops currently on the market are safe for human health and the environment. 9. Organic agriculture has solved all agricultural problems. We don't need GE. Organic farming seeks to maximize the health of the environment, the farmer, and the consumer. Organic farming came about as a response to the environmental and health problems associated with overuse of chemicals on conventional farms. Genetic engineering has contributed to this goal by reducing pesticide use (see #1). There are certain environmental or disease problems with no organic or conventional solution. There's nothing to make plants resistant to certain viruses, for example. Flood, drought, frost and salt tolerance have also been developed through GE and are now being tested. 10. We have already seen what is scientifically possible and it is not impressive. Every time a GE crop has been approved, farmers have embraced it and the GE acreage for each crop has quickly grown to 50 to 90 percent of the total acreage. To date most of the GE production is soybeans, corn, and cotton carrying two traits (herbicide tolerance and insect resistance). With unprecedented discoveries in genetics, many more traits are in the pipeline. |
| Which baby do you want? A dilemma for the 21st century parent-to-be [Genetic Future] Posted: 07 Sep 2008 05:56 AM CDT
It's worth reading through in full, but this statement from Susannah Baruch at Johns Hopkins caught my eye: There's speculation that people will have designer babies, but I don't think the data are there to support that. The spectre of people wanting the perfect child is based on a false premise. No single gene predicts blondness or thinness or height or whatever the 'perfect baby' looks like. You might find genetic contributors but there are so many environmental factors too.I'm unsure how selecting amongst these embryos doesn't count as making "designer babies" (it's still a choice, even if it's between a set of imperfect options), but I think Baruch's second paragraph is spot-on. It's clear from recent genome-wide association studies (GWAS) for height and weight that many (if not most) traits are hideously complex at the genetic level - a mess of common and rare genetic variants scattered throughout the genome, each contributing only the tiniest proportion of the overall variation. Height is a great example: GWAS results from more than 30,000 individuals have uncovered dozens of contributing variants, which together predict less than 5% of the variation in height. There's no doubt we'll uncover much of the remaining variation with emerging technologies (analysis of structural variation, and large-scale sequencing) and larger cohorts, by which time we'll likely have hundreds of contributing variants. The same will hold more or less true for other complex traits, including susceptibility to common diseases like diabetes or hypertension. The point is not that we will never understand the genetic basis of complex traits - we will, at least to a pretty good approximation, given advanced tools and sufficiently large cohorts. The point is that even once we understand the genetics of complex traits perfectly, that won't be enough to generate a "perfect baby" through embryo screening alone. To illustrate this, imagine - ten or fifteen years from now - a couple who have just had IVF to generate perhaps two dozen embryos, and want to use genetic testing to decide which one(s) to implant. There won't be a single, stand-out embryo, perfect and disease-free, because generating a "perfect" embryo - one with the "desirable" variant at every single position in the genome - runs up against a pretty serious probabilistic challenge. Let's say there are only 5,000 DNA variants that negatively affected human health (an under-estimate) each with a frequency of just 1%: that means you would get a "perfect" embryo around once in every 1022 attempts (that's a 1 followed by 22 zeroes, a stupidly large number). It's likely that methods to generate large numbers of embryos will be developed, particularly once stem cell technology enables sperm and egg cells to be created from adult tissue, but generating and screening 1022 embryos is no mean feat: at the rate of one every second, this would take you about 200,000,000,000,000 years, ten thousand times longer than the current age of the universe. So it's safe to say that there will be no perfect baby. Instead, the prospective parents will face a tough choice between embryo A, who will likely be tall, slim, smart and cancer-free but have a higher-than-average chance of bipolar, early-onset dementia, and infertility; embryo B, who will be a little shorter, dark-haired, probably fairly gregarious, resistant to coronary artery disease, susceptible to bowel cancer, hypertension and early deafness; embryo C, who will be of average intelligence, unlikely to suffer premature baldness, prone to mild obesity and diabetes, but not at a high risk of any of the other major common diseases; and embryos D-N, who present a similar panel of competing probabilities. Of course, a few embryos may carry mutations with known, severe health outcomes - those associated with rare diseases like muscular dystrophy, for instance. Embryo screening will have a very real impact on the frequencies of these conditions, just as we are already seeing following pre-natal diagnosis of conditions such as Down syndrome. But for the complex, common diseases there will be no easy answers; just a set of trade-offs. The parents-to-be will sit down together with dossiers listing a huge set of statistical predictions for each of their potential children, and make a decision as to which (if any) of these abstract collections of traits and risks they wish to bring into this world. Decisions don't get much more emotionally traumatic than this: not only will they be making a decision that will shape their own lives and that of their future offspring, parents will carry a new, extra burden of responsibility for the fate of their children. If they decide on embryo A, and their child goes on to develop severe bipolar disease, they will carry the guilt of that decision in addition to the trauma of the disease itself. That's not to say that embryo selection is unworkable - in fact, I think it's inevitable - but rather that this process is likely to require a degree of agonising trade-offs on the part of parents-to-be that is seldom fully appreciated. While I have no moral problem with the notion of embryo selection, part of me is glad that my child-bearing years are likely to be over before I have the chance to face this particular dilemma...  Subscribe to Genetic Future. Subscribe to Genetic Future. |
| Venter's exome, and the challenge of rare variants for personal genomics [Genetic Future] Posted: 07 Sep 2008 05:56 AM CDT  A team led by J. Craig Venter from the J. Craig Venter Institute has just published another paper on J. Craig Venter's favourite topic: J. Craig Venter. A team led by J. Craig Venter from the J. Craig Venter Institute has just published another paper on J. Craig Venter's favourite topic: J. Craig Venter.This study follows up on last year's publication of the complete sequence of Venter's genome, this time reporting a detailed analysis of a small but quite informative fraction of the genome: the exome, which consists of all of the pieces of DNA (called exons) that directly code for protein molecules. The exome is a favoured target of geneticists. There are two major reasons for this: firstly, the exome is enriched for functional sequence, whereas non-coding DNA has a much higher fraction of non-functional junk; and secondly, we understand protein-coding DNA much better than we do non-coding DNA. If a novel mutation alters a protein sequence, we have algorithms that can predict (with moderate accuracy) how likely it is to alter the function of the cell. In contrast, for most mutations in non-coding DNA we have almost no way to predict whether they are functional or not. So, like the drunkard looking for his keys under the lamp-post because the light is better there, geneticists are inclined to look hardest at the regions where they actually have some chance of finding something they can understand. Venter's mutations The article (which is open access, so you can read it yourself) has a number of interesting factoids about Venter's protein-coding genome that are highly relevant to personal genomics:
Even if a gene is known to be involved in disease, it is difficult to understand if a variant in the gene will have a phenotypic effect. We found that 99% of the [protein-altering variants] in disease genes could not be characterized by current literature. Different mutations in the same gene can cause different phenotypic effects [49], thus making it difficult to interpret possible phenotypes. Furthermore, some variants have phenotypic effects only under certain environments (see SOD2 and BDNF in Table 2 and [48]). Also, when looking at complex phenotypes, multiple variants in coding and non-coding regions are likely to be involved [63]–[66]. This genetic complexity, as well as exposure to various environmental factors, will need to be taken into account in assessing risk for various diseases.In other words, it will be quite some time before we can use a genome sequence to make realistic predictions about overall health (except for the unlucky few who carry mutations unambiguously associated with disease, such as a CAG repeat expansion in the HTT gene - in which case the predictions will tend to be dire). The next few years will be interesting times indeed for personal genomics companies, as their ability to generate oodles of genetic data with cheap sequencing increases exponentially faster than their capacity to explain what the data actually mean. The challenge of rare variants I want to draw particular attention to the implications of point 4 above (the fact that rare mutations are the most likely to alter protein function, and thus to have an effect on disease risk). The evolutionary basis for this association is trivially clear: if a variant has a serious negative effect on health then in most cases natural selection will keep it at a low frequency in the population, since really sick people tend to have fewer kids. Disease-causing variants can reach high frequencies under certain conditions (if they also provide benefits under certain situations, or if the disease only hits its victims after they've already reproduced, for instance) but all else being equal, evolution's scythe means that you're far more likely to find disease-causing variants at the rare rather than the common end of the spectrum. The reason this is so problematic is that rare disease-causing variants are also the hardest to find and characterise. I've mentioned a few times that the current crop of genome-wide association studies (GWAS), while reasonably well-powered to detect common disease-causing variants, have virtually no ability to find rare causal variants - even if these variants explain the majority of disease risk. This probably goes some way to explaining why even massive GWAS are capturing only a small proportion of the overall genetic risk for most common diseases. This arises primarily because the chips used in current GWAS only efficiently "tag" common variants. However, even once this technological barrier is lifted it will still be fiendishly difficult to assign function to rare variants: because there will be many millions of these variants, each at a low frequency, the sample sizes required to find those few associated with disease risk will be mind-bogglingly large - we're talking cohorts of millions of people, all with large-scale sequence data and well-collected information on environment and health. I have no doubt such studies will eventually be done, but it will take many years before we see the results. And of course, even with such massive cohorts, the rarest variants (those restricted to single families, or even just a few isolated individuals) will still slip through the statistical cracks - but such variants may well be the most important features in the genome sequence of any given individual, the ones disrupting that crucial tumour-suppressor gene or messing with neurotransmitter expression levels. If you have one of these nasty variants, you'll want to know about it, and you'll want to know what it does.  Beyond genetics Beyond geneticsUltimately, geneticists will have to deal with such variants using non-genetic methods. For instance, for many genes it may eventually be possible to create experimental assays that allow researchers to rapidly test whether a novel variant disrupts protein function; the mouse embryonic stem cell assays that can be used to test novel variants in the breast cancer gene BRCA2 are a proof of principle, as well as a demonstration of just how challenging this process will be. More broadly and ambitiously, we need to build and refine models of how human beings operate at a molecular level, integrating data from many fields of biology. If we understand which proteins interact within which cells, how these interactions influence protein dynamics, and where the binding sites for each interaction lie, we will have a much better chance of inferring the effect of an isolated change in protein sequence on overall cellular function and thus human health. Moving beyond the exome into non-coding DNA will require even more subtle and complex models including protein-DNA binding, the regulation of DNA modification and conformation, and the effects of non-coding RNA. In other words, ultimate personal genomics - the extraction of every byte of useful predictive information out of an individual's genome sequence - will require nothing less than an atomic-level understanding of the operation of the human machine. Now that is an effort I'd like to see Google throw its weight behind... (Venter image from Wikimedia Commons.) Ng, P.C., Levy, S., Huang, J., Stockwell, T.B., Walenz, B.P., Li, K., Axelrod, N., Busam, D.A., Strausberg, R.L., Venter, J.C., Schork, N.J. (2008). Genetic Variation in an Individual Human Exome. PLoS Genetics, 4(8), e1000160. DOI: 10.1371/journal.pgen.1000160 Subscribe to Genetic Future. |
| The challenges of psychiatric genetics [Genetic Future] Posted: 07 Sep 2008 05:52 AM CDT  Back in April I posted on the elusive genetics of bipolar disorder, a crippling psychiatric condition affecting over 2% of the population in any given year. Back in April I posted on the elusive genetics of bipolar disorder, a crippling psychiatric condition affecting over 2% of the population in any given year.The major message from that article is that although bipolar disorder is massively influenced by genetic factors (around 85% of the variation in risk is thought to be due to genetics) we still don't really have the faintest idea exactly which genes are involved. This is despite three reasonably large genome-wide association studies involving over 4,000 bipolar patients in total, which generated weak and contradictory results and failed to provide a single compelling candidate for genetic variation underlying this disease. This disappointing result has also held largely true for other psychiatric conditions with strong genetic components, such as schizophrenia, major depression and autism. Genetic studies of these conditions have had some success identifying rare mutations that underlie severe cases, but the vast majority of the genetic variants contributing to risk remain undiscovered. There are several reasons why genome-wide association studies can fail to yield significant harvests of disease-associated genes. I summed these up with respect to bipolar disease as follows: The researchers are surely hoping that small effect sizes are the major problem, since this is the easiest problem to remedy (simply increase sample sizes). Disease heterogeneity - in other words, multiple diseases with distinct causes that all converge on a bipolar end-point - also seems like a particularly plausible explanation given the complexities of mental illness. It's also likely that various types of genetic variants that are largely invisible to existing SNP chips, like rare variants and copy-number variation, are important.The same story probably holds largely true for other psychiatric conditions. In this week's issue of Nature, a news article and an editorial both tackle the challenges of psychiatric genetics, and lay out the ambitious strategies currently being pursued by researchers around the world to overcome them. Small effect sizes The first hurdle that I describe above is the fact that most of the variants underlying these conditions probably have very small effect sizes (only increasing risk by less than 20%). Such variants will only be identified by cranking up sample sizes immensely, an approach that has yielded some limited success for other genetically complex traits such as height and obesity. The Nature news feature has a table listing some of the major collaborative efforts currently collecting genetic information from the very large cohorts required to dissect out the basis of these conditions: 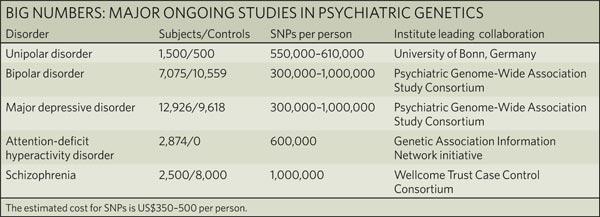 In most cases, these samples are being built up by pooling results from multiple different studies, often gathered by groups from around the world. As sample sizes increase the power of studies to detect small-effect variants grows. The effect of sample size on the power of genome-wide association studies is illustrated in the graph below from a recent review by Peter Visscher*: 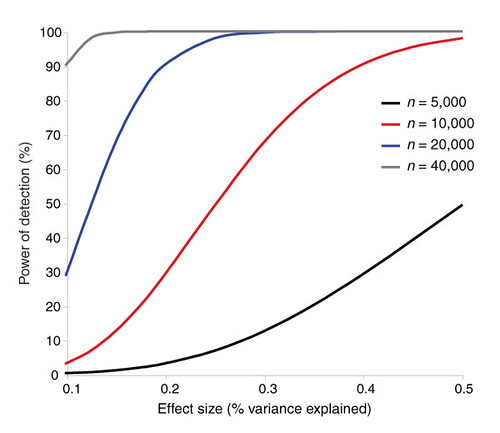 Take a single genetic variant that explains just 0.5% of the variance in the risk of a psychiatric disorder. With a sample size of 5,000 individuals with that disorder you still have a mere 50% chance of detecting that variant. Double your sample size, and that probability jumps to a near-certainty of detection - and your power of detecting even smaller-effect variants (explaining, say, 0.2% of the risk) starts to climb to respectable levels. By staring at those curves for a while, and bearing in mind that many of the variants found by recent genome-wide association studies explain well under 0.2% of the risk variance, you will quickly start to appreciate why researchers are pushing for ever-larger disease populations to work with. With truly enormous samples on the order of 50 to 100 thousand patients - not out of the question for international consortiums studying reasonably common diseases such as bipolar - the power to detect even very weak risk variants becomes reasonable. If there are common genetic variants contributing to the risk of these diseases, such large collaborative studies will eventually find them; so long, of course, as they can tackle the next (and potentially far more serious) problem of disease heterogeneity. Complex, heterogeneous diseases  The second major problem I mentioned with analysing the genetic basis of these diseases is that they are complex, multifactorial, and extremely difficult to diagnose and classify. Psychiatric conditions are probably the most difficult area of medicine to draw hard boundaries: many symptoms are shared by multiple conditions, and many patients display a diffuse constellation of clinical signs that makes a clean diagnosis impossible. The second major problem I mentioned with analysing the genetic basis of these diseases is that they are complex, multifactorial, and extremely difficult to diagnose and classify. Psychiatric conditions are probably the most difficult area of medicine to draw hard boundaries: many symptoms are shared by multiple conditions, and many patients display a diffuse constellation of clinical signs that makes a clean diagnosis impossible.This complexity and heterogeneity is the basis of considerable tension between geneticists and neuroscientists, which is explored in the Nature editorial. Basically, to build up those massive sample sizes shown above geneticists are forced to lump together patients with a variety of clinical symptoms, thus essentially ignoring the complexity inherent in these conditions - a failure that neuroscientists find inexcusable. In turn, geneticists (like myself) get seriously annoyed by the tendency of neuroscientists to make big, bold claims about disease mechanisms based on studies with tiny sample sizes. Both sides make reasonable criticisms. As I said in the quote from my previous article above, it seems likely that disease heterogeneity - that is, multiple diseases states with the same broad end point being simplistically lumped together - plays a major role in the failure of genome-wide association studies of psychiatric conditions; at the same time, the scientific value of much of the "sexy" neurobiology currently being published (e.g. functional MRI finds that conservatives have lower activity in "compassion" centres of the brain, or whatever) is sometimes highly questionable. Both sides of this scuffle have something to learn from their opponents. The editorial argues, sensibly, that geneticists and neuroscientists just need to start getting along. The ideal situation is one in which rigorous clinical assessments are used to generate patient cohorts that are as homogeneous as possible that can then be subjected to large-scale genetic analysis. One especially promising avenue is the use of "endophenotypes" - that is, simple and easily quantifiable traits that are sometimes but not always associated with a particular disease. Cleanly defined endophenotypes, such as very specific dysfunctions of brain activity, may prove much more amenable to genetic dissection than the larger, more complex diseases they are associated with. Comprehensively tackling the genetic of psychiatric conditions will require a forceful and combined approach drawing on the clinical expertise of neuropsychiatrists and the experience of geneticists in unravelling the genetic mechanisms of complex traits. To some extent this is happening already (no large genetics consortium would be naive enough to embark on a multi-million dollar project without consulting clinical experts) - but obviously there is considerable room for improvement. Moving beyond common SNPs  Current genome-wide association studies currently rely largely on the use of single-letter variations in DNA called single nucleotide polymorphisms (SNPs), mainly because these are easy to analyse and can be simultaneously analysed in their hundreds of thousands using chip-based assays. For various reasons almost all of the SNPs on current genome-wide association chips are common sites of variation, present at a frequency of 5% or more in the population. However, recent studies have made it look increasingly likely that a large proportion of the genetic risk of common diseases lies in types of genetic variation that cannot be detected using common SNPs: rare variants, and large-scale rearrangements of DNA known as structural variation. Current genome-wide association studies currently rely largely on the use of single-letter variations in DNA called single nucleotide polymorphisms (SNPs), mainly because these are easy to analyse and can be simultaneously analysed in their hundreds of thousands using chip-based assays. For various reasons almost all of the SNPs on current genome-wide association chips are common sites of variation, present at a frequency of 5% or more in the population. However, recent studies have made it look increasingly likely that a large proportion of the genetic risk of common diseases lies in types of genetic variation that cannot be detected using common SNPs: rare variants, and large-scale rearrangements of DNA known as structural variation.The approaches required to capture these variants are already pretty well-known, although they remain expensive and technically challenging. In an ideal world, genome-wide association studies would be truly genome-wide - in other words, they would utilise the entire DNA sequence of all of the patients and controls in the sample to find every possible genetic variant that might contribute to disease. Unfortunately, such an approach is currently out of reach, for several reasons:
Both approaches have their limitations. The success of the candidate gene approach will be constrained by researchers' ability to identify the genes most likely to be involved in a particular disease - but in fact our currently severaly limited understanding of disease genetics is precisely why we need to study this issue in the first place! (In the Nature news piece, Harvard's Steven Hyman memorably describes this approach as "like packing your own lunch box and then looking in the box to see what's in it.") And while chip-based detection of structural variation is rapidly increasing in resolution, it's extremely difficult to determine which of the variants identified in a study are disease-causing and which are harmless polymorphisms - this is currently done probabilistically, by showing that there is an enrichment of new variants in disease cases compared to controls, but this approach cannot tell you which of the identified variants are actually causative. From psychiatric genetics to genetic psychiatry? There are several important reasons researchers are interested in the genetics of mental illness: identifying causal genes helps to dissect out the molecular pathways involved in disease, and may help to pull out otherwise invisible sub-types of a disease; studying "extreme" mental phenotypes may illuminate the genetic basis of variation in cognition and personality traits in "normal" people; and, perhaps most importantly, by identifying the genes underlying psychiatric diseases we may be able to target at-risk individuals for monitoring and intervention, potentially heading off severe disease before it takes hold. In the headlong pursuit of these goals the field of psychiatric genetics has developed an unfortunate reputation built on bold claims made with limited evidence, and literally hundreds of reported associations that have completely failed to stand up to replication. Just a couple of years ago the shiny new tools of large-scale genomics promised an end to this ignoble period in the history of the field; unfortunately, the introduction of larger samples, higher genomic coverage and increased statistical rigour has not brought the desired clarity to the field, but rather seems to have increased the levels of confusion and uncertainty. If anything, that crucial third goal - using genetic to predict the risk of mental illness - now appears further away than it did just a couple of years ago. Back in early 2007 we didn't have many convincing genetic predictors of mental illness, but at least it was possible to imagine that emerging genomic technologies might identify a small core set of large-effect variants that would help clinicians to predict disease risk. Right now we still don't have many useful genetic predictors, and that illusion of hope is gone. In summary: while there's no doubt that these conditions do have a strong genetic basis, it's now abundantly clear that this basis is frighteningly complex, with common variants of moderate-to-large effect - the types of variants that would be most useful for risk prediction - being essentially absent. It's going to take many years, massive cohorts, the clever application of new genomic technologies, and a willingness from both neuroscientists and geneticists to listen to one another to move this field forward. (Brain scan image from Science Photo Library.) * Thanks to reader Chris for providing me with the citation, which I had carelessly misplaced! Subscribe to Genetic Future. |
| How well does your genome predict your postcode? [Genetic Future] Posted: 07 Sep 2008 05:52 AM CDT Well, it's far from GPS precision, but the concordance between this genetic map of Europe (below left) and the physical sampling locations of populations throughout Europe (below right) is pretty good for a first draft:
Dienekes has an excellent discussion of the technical details, while Razib has labelled a plot showing all of the individuals in the study to make it easier to assess the degree of scatter and overlap. The take-home message: rather than being one homogeneous mass, Europeans in fact show considerable population substructure, such that genetic information can be used to roughly predict geographical ancestry. An analysis of just a few hundred thousand genetic markers (i.e. less than is currently offered by personal genomics companies 23andMe or deCODEme) would be more than adequate in most cases to distinguish a Pole from a Parisian, or a Swede from a Spaniard. (To be more precise, it would be sufficient to discriminate between individuals for whom most ancestors were natives of these regions; recent migrants will obviously be misclassified.) What drove these genetic differences? Mostly it will have been chance - random increases or decreases in the frequency of markers throughout the genome accumulated over a few millennia of genetic isolation. But at least some of these differences have been driven by natural selection: for instance, the lactase gene LCT, which has been subject to strong selection to allow lactose digestion in adults in populations reliant on dairy agriculture, represents 9 out of the top 20 most differentiated markers; a marker in the gene HERC2, which is associated with eye colour variation and has been under selection in Europeans and Asians, comes in at number 19. This indicates that at least some of the genetic - and thus physical and possibly behavioural - differences between the various European populations stem from evolutionary adaptation to their local environments. I'll leave the technical commentary to Dienekes, but I do want to make one important point: the accuracy of the map will have been limited by the fact that the markers used in this study represent sites of common variation; data from large-scale genome sequencing will generate far, far better maps. The major reason for this is that sequencing will provide information on rare, highly spatially-restricted variants - many of which will be limited to single families and thus be extremely informative about geographical ancestry. Basically, if you had complete genome sequences from enough Europeans you could reconstruct the genetic map of Europe with exquisite precision. In addition to empowering genetic genealogists, researchers could use deviations between the genetic and physical maps to make powerful inferences about historical migration events and recent episodes of natural selection. With any luck, this is the sort of data that will simply fall out from large-scale population genomic studies being conducted over the next decade or so. Update: Kambiz at Anthropology.net puts these results in a broader scientific context. Lao et al. (2008). Correlation between Genetic and Geographic Structure in Europe. Current Biology DOI: 10.1016/j.cub.2008.07.049 Image source: Figure 1 from Lao et al. Subscribe to Genetic Future. |
| The gene for Jamaican sprinting success? No, not really. [Genetic Future] Posted: 07 Sep 2008 05:49 AM CDT  Anyone who has walked past a TV set over the last few days will have seen footage of the remarkable Jamaican sprinter Usain Bolt, who comfortably cruised to victory (and a world record) in the Olympic 100 metre sprint, and as I write this has just done precisely the same thing in the 200 metre sprint. The interest in Bolt stems not from the fact that he wins his races, but rather from the contemptuous ease with which he does so. Anyone who has walked past a TV set over the last few days will have seen footage of the remarkable Jamaican sprinter Usain Bolt, who comfortably cruised to victory (and a world record) in the Olympic 100 metre sprint, and as I write this has just done precisely the same thing in the 200 metre sprint. The interest in Bolt stems not from the fact that he wins his races, but rather from the contemptuous ease with which he does so.And Bolt is not the only Jamaican to impress in short distance events in Beijing: the country's women's sprint team took all three medals in their 100 metre dash. Naturally, these performances have provoked widespread speculation about the basis of Jamaica's sprinting success, and the short-distance prowess of other populations of West African ancestry. One controversial suggestion has drawn the most headlines: that sprinting is in their genes, or rather in one gene in particular - variously referred to as "Actinen A" or "ACTN3". This gene has been the subject of a recent rash of news stories sparked by Bolt's victories, all of which refer to comments by Jamaican academic Errol Morrison in the Jamaica Gleaner over a month ago. The Gleaner article summarised the (unpublished) results of a collaboration between Morrison and a group at the University of Glasgow: At the base of sprint speed are the fast-twitch muscle fibres stocked with the speed protein Actinen A. And early data indicate that 70 per cent of Jamaican athletes have the gene for Actinen A. Only 30 per cent of Australian athletes studied had the gene.(The Gleaner reporter, Martin Henry, astonishingly went on to speculate that this gene may help to explain why Jamaicans are "also disproportionately aggressive and violent".) The Daily Mail followed up on the story two weeks later with a marginally more coherent account: What they have found - and Morrison emphasises the findings are preliminary - is that fast men have a special component called Actinen A in their fast-twitch muscles, which determine whether humans are sprinters or plodders. It is found in 70 per cent of Jamaicans. In a control study of Australians, only 30 per cent were found with it.The "preliminary" nature of the findings didn't stop the Daily Mail reporter from following this paragraph with the conclusion that this result "would seem to explain why Jamaicans punch above their weight among sprinters". Similarly definitive statements were made by other reporters continuing the story after Bolt's 100 metre victory; one rare exception was a fairly well-balanced piece in Slate. The stories take advantage of a widespread perception - by no means totally unjustified, but nonetheless controversial - that Jamaicans and other groups of West African ancestry have a genetic advantage when it comes to raw muscle power. Having apparent scientific evidence to support this perception is a reporter's dream; the headlines write themselves. So, how good is this scientific evidence? Does the "Actinen A" gene (whatever that is) actually influence sprinting performance? And if so, does it explain the difference in explosive power between Jamaicans and the rest of the world? The answers, as it turns out, are "probably" and "not really". The ACTN3 gene and muscle performance At this point I probably should confess to having a more than casual interest in this story: I was one of the authors on the first study showing an association between this gene and elite athlete status back in 2003, and this gene has been the central focus of my research for a good part of the last six years. (The opinions I express here are purely my own, by the way, and in no way are meant to represent the views of my research institute.) The ACTN3 gene encodes a protein called α-actinin-3 ("Actinen A" is a misnomer of uncertain origin propagated by lazy reporters), which is found within the fast fibres of muscle - the cells that are required for generating rapid, forceful contraction in activities such as sprinting and weightlifting. Interestingly, the human ACTN3 gene comes in two forms in the general population: there's a normal, functional version called 577R, and a "defective" version called 577X, which contains a single base change that prevents the production of α-actinin-3. People who have two copies of the 577X version (I'll refer to them as X/X) produce absolutely no α-actinin-3 in their fast muscle fibres. These people don't suffer from muscle disease as a result of this deficiency - in fact, there's a pretty good chance that you're one of them. The frequency of the 577X variant differs around the world, but overall somewhere between one-sixth and one-quarter of the world's population (at least a billion people worldwide) are X/X, and therefore completely deficient in α-actinin-3. So lack of α-actinin-3 clearly doesn't destroy your muscle; however, over the last five years we and other groups have assembled evidence suggesting that it does influence how good your muscle is at generating explosive power. We first showed in 2003 that X/X individuals are significantly under-represented among elite Australian sprint/power athletes, suggesting that the absence of α-actinin-3 in X/X individuals is detrimental to optimal muscle power generation. This association has since been replicated in four separate athlete studies by groups in Europe and the US; there is also weaker but reasonably consistent evidence that α-actinin-3 deficiency results in slightly higher endurance capacity, both in human athletes and in a mouse model generated by our group. In addition, several groups have reported that X/X individuals in the general population display lower muscle strength and reduced sprint performance. Importantly, the latter two studies suggest that the proportion of the variance in strength and sprint performance in the general population explained by the ACTN3 variant is around 2-3%. So for most of us lazy slobs this gene has a pretty trivial effect - almost completely drowned out by noise from the effects of diet, exercise levels and other genes. (Certainly there are dozens or even hundreds of other genes influencing physical performance, some of which - like the ACE gene - have been fairly well-studied, but most of which are completely unknown and uncharacterised; and environmental factors play about as large a role as genes do in traits like muscle strength and cardiorespiratory performance.) However, even 2-3% can make a striking difference at the very elite level: of the 51 Olympic-level sprint/power athletes analysed in our original study and a follow-up analysis in Greek athletes not a single individual was X/X (compared to about 10 expected). In fact, X/X Olympian sprint athletes are unusual enough that identifying a single Spanish Olympic short-distance hurdler with α-actinin-3 deficiency was enough to warrant its own publication. So the absence of α-actinin-3 means very little to most of us, but to a young athlete craving 100 metre Olympic superstardom it could make all the difference in the world. The same could be said of many other genetic variants, of course; Olympic sprinters, essentially, are those unlikely individuals at the vanishing edge of the probability distribution for whom nearly every genetic coin has come up heads. Does the ACTN3 gene explain Jamaican sprinting prowess? The underlying argument here is intuitively simple: (1) variation in the ACTN3 gene is strongly associated with elite sprint athlete status; (2) the "sprint" version of ACTN3 is more common in Jamaicans than in individuals of European ancestry; therefore (3) this variant may well play a role in the increased sprinting prowess of Jamaicans relative to Europeans. At first blush this sounds pretty convincing; however, while ACTN3 may play some role in the disproportionate success of Jamaican sprinters, I'd argue that it's likely to be a pretty small one. Here's why:
Beyond "the gene for speed" I'm certainly not arguing here that genetics doesn't play any role in Bolt's success - or in the remarkable over-representation of West African descendents in Olympic short-distance track events, or the similarly impressive skew towards East Africans among marathon runners. In fact I think most geneticists would be staggered if this was the case, even though direct evidence for underlying genes is currently very thin on the ground. Rather, my point is that an excessive emphasis on ACTN3 as a major explanation for Jamaican success does a grave disservice to the complex interplay of genetic and environmental factors required for top-level athletic performance. This suggestion goes against everything we've learnt about the genetics of complex traits from recent genome-wide association studies, which have revealed that quantitative traits (like height and body weight) are frequently influenced by dozens to hundreds of genes, each of small effect; if anything, it's likely that athletic performance will be even more genetically complex than these traits. The ACTN3-centred argument also dismisses the importance of Jamaica's impressive investment in the infrastructure and training system required to identify and nurture elite track athletes, the effects of a culture that idolises local track heroes, and the powerful desire of young Jamaicans to use athletic success to lift themselves and their families out of poverty. It is almost certainly true that Usain Bolt carries at least one of the "sprint" variants of the ACTN3 gene, but then so do I (along with around five billion other humans worldwide). Indeed, I'm fortunate enough to be lugging around two "sprint" copies - but that doesn't mean you'll see me in the 100 metre final in London in 2012. Unfortunately for me, it takes a lot more than one lucky gene to create an Olympian. (Image: Phil McElhinney.) Subscribe to Genetic Future. |
| Genetic Future is moving, and so am I [Genetic Future] Posted: 07 Sep 2008 05:49 AM CDT Genetic Future is moving to a shiny new home at ScienceBlogs. This domain will remain as an archive site, but for fresh content you will need to update your links as follows: New URL: http://scienceblogs.com/geneticfuture/ New RSS feed: http://feeds.feedburner.com/scienceblogs/geneticfuture Some of you familiar with the ScienceBlogs network might be wondering if this move heralds a transition into left-wing political blogging, but don't worry: my articles will continue to be focused on reporting advances in human genomics and critiquing the genetic testing industry. Just a few weeks after the transition I'll also be physically moving from Sydney to a new life in Cambridge, UK. Posting on the new site will be light during this move and regulars will notice a few recycled posts to fill in the awkward silences, but bear with me - in a couple of weeks there will be plenty of fresh human genetics goodness. Hope to see you all at the new domain, Daniel. Subscribe to Genetic Future. |
| BREAKING NEWS [Genetic Future] Posted: 07 Sep 2008 05:48 AM CDT Hopefully I now have the attention of at least a small proportion of my RSS subscribers; here's a friendly reminder: Genetic Future has moved and you need to update your RSS feed by clicking HERE. This feed will be inactivated shortly, and this domain will become an archive site. Daniel. Subscribe to Genetic Future. |


| You are subscribed to email updates from The DNA Network To stop receiving these emails, you may unsubscribe now. | Email Delivery powered by FeedBurner |
| Inbox too full? Subscribe to the feed version of The DNA Network in a feed reader. | |
| If you prefer to unsubscribe via postal mail, write to: The DNA Network, c/o FeedBurner, 20 W Kinzie, 9th Floor, Chicago IL USA 60610 | |
No comments:
Post a Comment